我在 R 上有一個類似于這個的資料框,只有 2000 行長。在整個資料框中,我在稱為“id read”的單個讀取中具有 SEQ1 和 SEQ2 的這種交替。這些序列交替出現,SEQ1總是與SEQ1相距1個核苷酸,而SEQ2與SEQ1相距約335個核苷酸,有時會跳躍到670個。從末端坐標的值可以看出,序列既有正向,也有反向有時小于起始坐標。
| 順序 | 我害怕 | 開始 | 結尾 | 序列距離 | 序列長度 |
|---|---|---|---|---|---|
| 序列1 | 我害怕 | 90 | 105 | 1 | 15 |
| 序列2 | 我害怕 | 440 | 458 | 335 | 18 |
| 序列1 | 我害怕 | 459 | 474 | 1 | 15 |
| 序列2 | 我害怕 | 808 | 826 | 334 | 18 |
| 序列1 | 我害怕 | 827 | 812 | 1 | 15 |
| 序列2 | 我害怕 | 1148 | 1156 | 336 | 18 |
| 序列1 | 我害怕 | 1157 | 1172 | 1 | 15 |
| 序列2 | 我害怕 | 1850 | 1868年 | 678 | 18 |
| 序列1 | 我害怕 | 1869 | 1854年 | 1 | 15 |
| 序列2 | 我害怕 | 2187 | 2205 | 333 | 18 |
| 序列1 | 我害怕 | 2206 | 2221 | 1 | 15 |
| 序列2 | 我害怕 | 2887 | 2905 | 666 | 18 |
有人對如何繪制這些資料并直觀地顯示這些序列在讀取中的模式有任何想法嗎?我嘗試使用水平線、棒棒糖、點進行繪圖,但這些方法都不能有效地表示我擁有的資料量并直觀地理解這些序列的行為。有人知道如何繪制模式嗎???如果我愿意,我也可以只繪制我擁有的大型資料幀的一部分,但至少我想了解這些序列在考慮到超長讀取時的特殊性。
uj5u.com熱心網友回復:
我仍然不完全確定您在尋找什么,但如果每行i wheresequence == "SEQ"都有成對的行i 1where sequence == "SEQ2",您可以計算相對的開始和結束站點,然后嘗試將其可視化。
假設您的資料位于名為 的變數中df,您可以按如下方式計算這些資料。
df <- transform(
df,
rel_start = ifelse(
as.character(sequence) == "SEQ1",
start - start,
start - c(0, head(start, -1))
),
rel_end = ifelse(
as.character(sequence) == "SEQ1",
end - start,
end - c(0, head(start, -1))
)
)
然后對于可視化,您可以使用geom_segment(). 您可以使用箭頭來指示讀取的方向。
library(ggplot2)
ggplot(df, aes(rel_start, y = seq_along(start), colour = sequence))
geom_segment(aes(xend = rel_end, yend = seq_along(start)),
arrow = arrow(length = unit(2, "mm")))
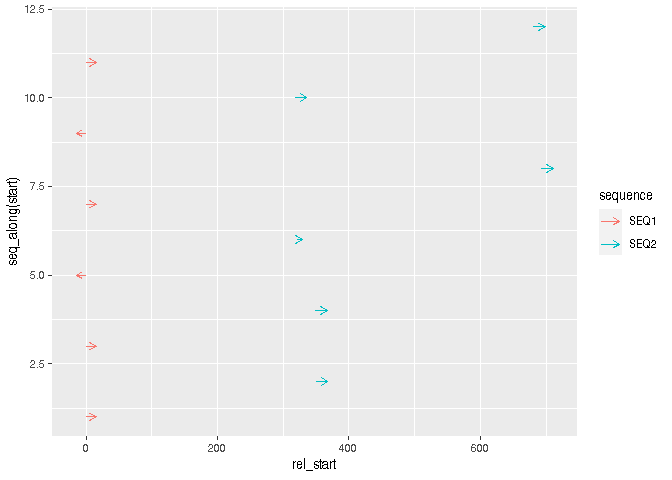
資料加載:
txt <- "sequence id read start end sequencedistance sequencelength
SEQ1 id read 90 105 1 15
SEQ2 id read 440 458 335 18
SEQ1 id read 459 474 1 15
SEQ2 id read 808 826 334 18
SEQ1 id read 827 812 1 15
SEQ2 id read 1148 1156 336 18
SEQ1 id read 1157 1172 1 15
SEQ2 id read 1850 1868 678 18
SEQ1 id read 1869 1854 1 15
SEQ2 id read 2187 2205 333 18
SEQ1 id read 2206 2221 1 15
SEQ2 id read 2887 2905 666 18"
df <- read.table(text = txt, header = TRUE)
轉載請註明出處,本文鏈接:https://www.uj5u.com/caozuo/445455.html