一個重要的倍訓:
機器學習-資料挖掘的流程(CRISP-DM):圍繞資料進行如下6個活動進行倍訓式地探索活動
- 商業理解
- 資料理解
- 資料準備
- 建立模型
- 模型評估
- 方案實施
一個重要的概念:
特征工程:最大限度地從原始資料中提取特征以供演算法和模型使用,包括如下幾個主要部分:
- 資料預處理:標準化、縮放、缺失、變換、編碼等
- 特征產生:結合業務資料、派生新的特征
- 特征選擇:通過各種統計量、模型評分等,篩選合適的特征
- 降維:PCA、LDA等減少特征個數
兩個重要的演算法:
決策樹:
- 構建決策樹
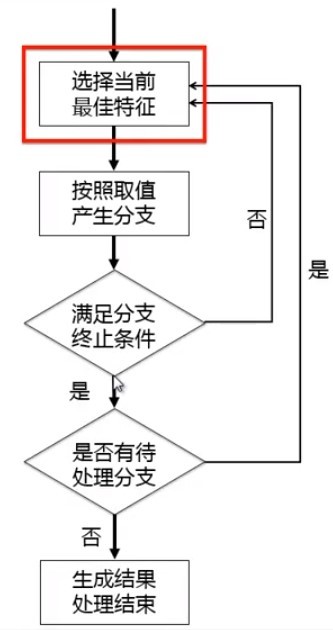
- 如何“選擇當前最佳特征”:
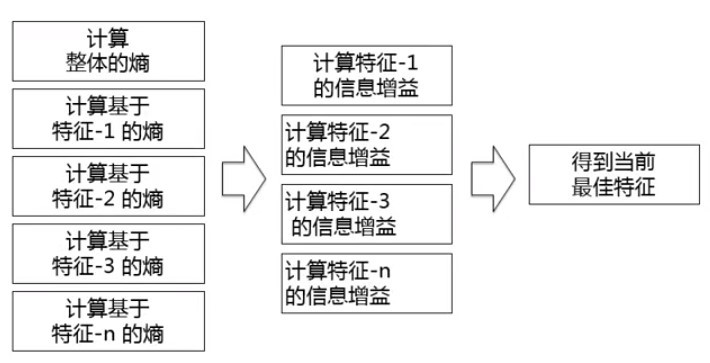
其中包含的主要概念:
資訊熵:資訊論理的概念,香農提出;描述混亂程度的度量;取值范圍:0~1(值越大,越混亂),計算公式如下:
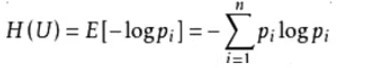
資訊增益:資訊是確定性的增加;從一個狀態到另一個狀態資訊的變化;資訊增益越大,對確定性貢獻越大,
- 決策樹演算法的主要分類:
- ID3系列(Iterative Dichotomiser 3, 迭代樹三代):核心是資訊熵,根據資訊增益決策樹的節點;存在一些問題:資訊度量不合理-傾向于選擇取值多的欄位;輸入型別單一 - 離散型;不做剪枝,容易過擬合,
- C4.5:和ID3相比的改進:用資訊增益率代替資訊增益;能對連續屬性進行離散化,對不完整資料進行處理;進行剪枝,
- C50:C4.5相比的改進:使用了boosting; 前修剪、后修剪
- CART(Classification and Regression Tree): 核心是基尼系數(Gini); 分類是二叉樹;支持連續值和離散值;后剪枝進行修剪;支持回歸,可以預測連續值
- 決策樹的具體施行:
-
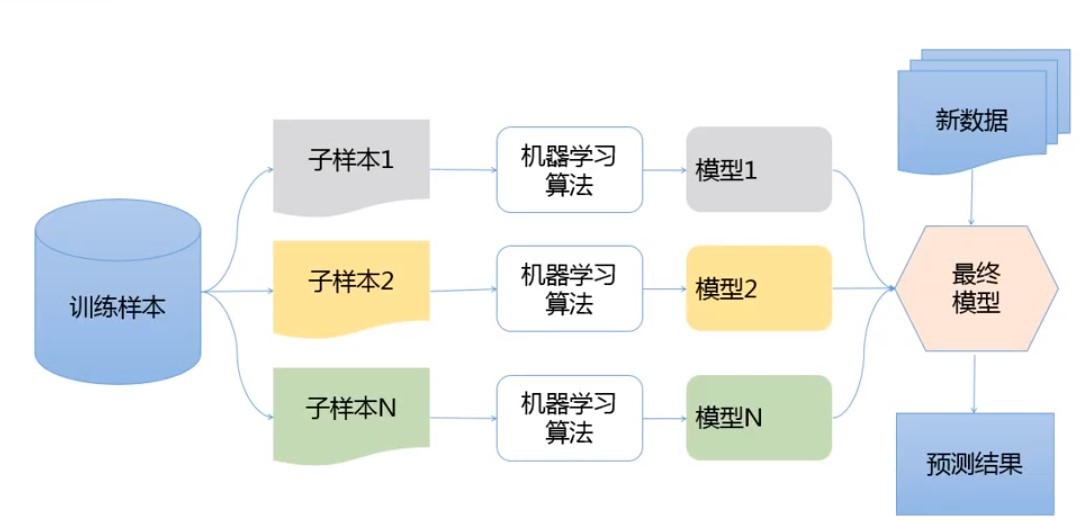
集成學習:針對同一資料集,訓練多種學習器,來解決同一問題,
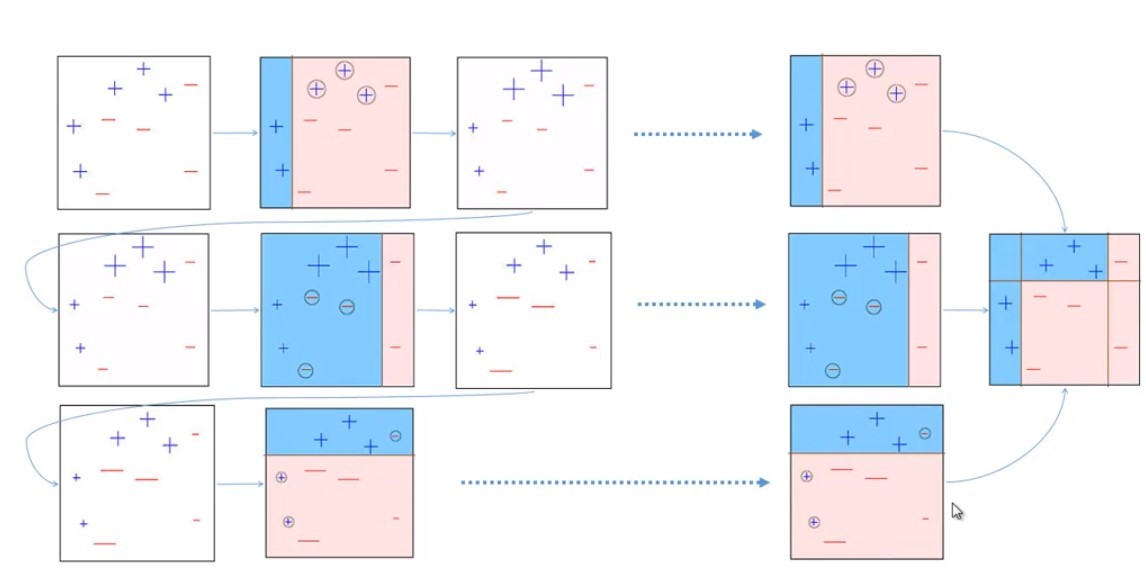
- Bagging: 有放回抽樣構建多個子集;訓練多個分類器;最終結果由各分類器結果投票得出;(實作非常簡單),
-
Boosting: 重復使用一類學習器來修改訓練集;每次訓練后根據結果調整樣本的權重;每個學習器加權后的線性組合即為最終結果,
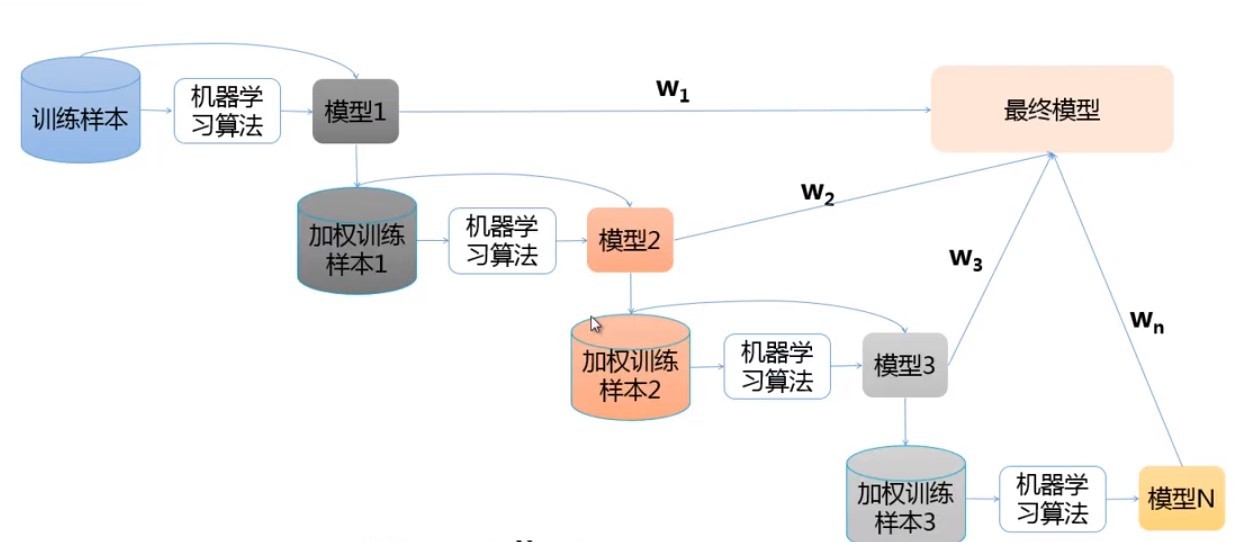
-
AdaBoost:
- 其他實作方法:
Stacking: 由兩級組成,第一級為初級學習器,第二級為高級學習器;第一級學習器的輸出作為第二級學習器的輸入,
隨機森林:
由許多決策樹組成,樹生成時采用了隨機的方法;Smart Bagging;生成步驟:1.隨機采樣,生成多個樣本集;2.對每個樣本集構建決策樹,
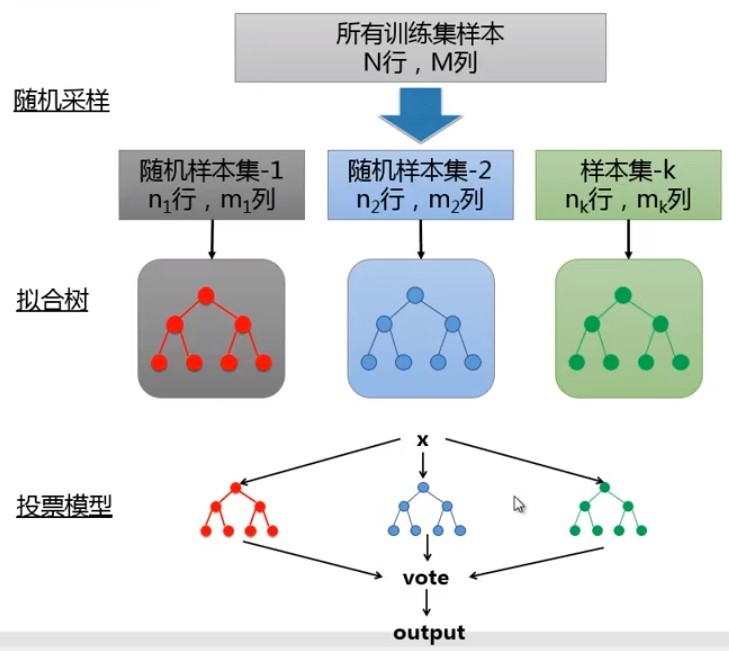
其優點包括:可以處理多分類;不會過擬合;容易實作并行;對資料集容錯能力強,
參考 - 1. R語言運用隨機森林的例子:
###################################################
### Gene (Feature) Selection 基因特征選擇: 1.過濾方法;2.封裝方法
###################################################
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(version = "3.11")
BiocManager::install(c("Biobase", "genefilter"))
BiocManager::install(c("ALL"))
library(Biobase)
library(ALL)
data(ALL)
ALLb <- ALL[,tgt.cases]
rowIQRs <- function(em)
rowQ(em,ceiling(0.75*ncol(em))) - rowQ(em,floor(0.25*ncol(em)))
plot(rowMedians(es),rowIQRs(es),
xlab='Median expression level',
ylab='IQR expression level',
main='Main Characteristics of Genes Expression Levels')
library(genefilter)
ALLb <- nsFilter(ALLb,
var.func=IQR,
var.cutoff=IQR(as.vector(es))/5,
feature.exclude="^AFFX")
ALLb <- ALLb$eset
es <- exprs(ALLb)
dim(es)
#ANOVA過濾
f <- Anova(ALLb$mol.bio,p=0.01)
ff <- filterfun(f)
selGenes <- genefilter(exprs(ALLb),ff)
sum(selGenes)
ALLb <- ALLb[selGenes,]
ALLb
es <- exprs(ALLb)
plot(rowMedians(es),rowIQRs(es),
xlab='Median expression level',
ylab='IQR expression level',
main='Distribution Properties of the Selected Genes')
# 用隨機森林(適合用于處理包含大量特征的問題)進行過濾:隨機森林由一組決策樹構成,取決于分析的問題采用回歸樹還是分類樹 - 每棵樹都是通過自助法抽樣(從原始資料集中用有放回抽樣法隨機抽取N個個案)進行訓練
# 對于回歸問題,采用每棵樹的預測值得平均值作為這些組合的預測值,對于分類問題,則采用投票機制
featureNames(ALLb) <- make.names(featureNames(ALLb))
es <- exprs(ALLb)
library(randomForest)
dt <- data.frame(t(es),Mut=ALLb$mol.bio)
rf <- randomForest(Mut ~ .,dt,importance=T)
imp <- importance(rf)
imp <- imp[,ncol(imp)-1]
rf.genes <- names(imp)[order(imp,decreasing=T)[1:30]]
sapply(rf.genes,function(g) tapply(dt[,g],dt$Mut,median))
library(lattice)
ordMut <- order(dt$Mut)
levelplot(as.matrix(dt[ordMut,rf.genes]),
aspect='fill', xlab='', ylab='',
scales=list(
x=list(
labels=c('+','-','*','|')[as.integer(dt$Mut[ordMut])],
cex=0.7,
tck=0)
),
main=paste(paste(c('"+"','"-"','"*"','"|"'),
levels(dt$Mut)
),
collapse='; '),
col.regions=colorRampPalette(c('white','orange','blue'))
)
#用特征聚類的組合進行過濾
library(Hmisc)
vc <- varclus(t(es))
clus30 <- cutree(vc$hclust,30)
table(clus30)
getVarsSet <- function(cluster,nvars=30,seed=NULL,verb=F)
{
if (!is.null(seed)) set.seed(seed)
cls <- cutree(cluster,nvars)
tots <- table(cls)
vars <- c()
vars <- sapply(1:nvars,function(clID)
{
if (!length(tots[clID])) stop('Empty cluster! (',clID,')')
x <- sample(1:tots[clID],1)
names(cls[cls==clID])[x]
})
if (verb) structure(vars,clusMemb=cls,clusTots=tots)
else vars
}
getVarsSet(vc$hclust)
2. R語言資料科學包串列 : https://www.cnblogs.com/yxmings/p/14213573.html
轉載請註明出處,本文鏈接:https://www.uj5u.com/houduan/243778.html
標籤:其他
上一篇:java的多載與重寫