我可以看到很多關于這個主題的帖子,但沒有一個解決這個問題。如果我錯過了相關答案,我們深表歉意。我有一個大型蛋白質表達資料集,樣本如下:rep1_0hr、rep1_16hr、rep1_24hr、rep1_48hr、rep1_72hr .....
和 2000 多種蛋白質。換句話說,每個樣本都是不同的發育時間點。
如果有任何興趣,原始資料集是pRolocdataR 中的包中的“mulvey2015”,我將其轉換為SummarizedExperimentRStudio 中的物件。
assay()我首先對資料(資料集的一個SummarizedExperiment,以獲得 12 個集群)運行 k-means 聚類:
k_mul <- kmeans(scale(assay(mul)), centers = 12, nstart = 10)
然后:
summary(k_mul)
產生了預期的輸出。
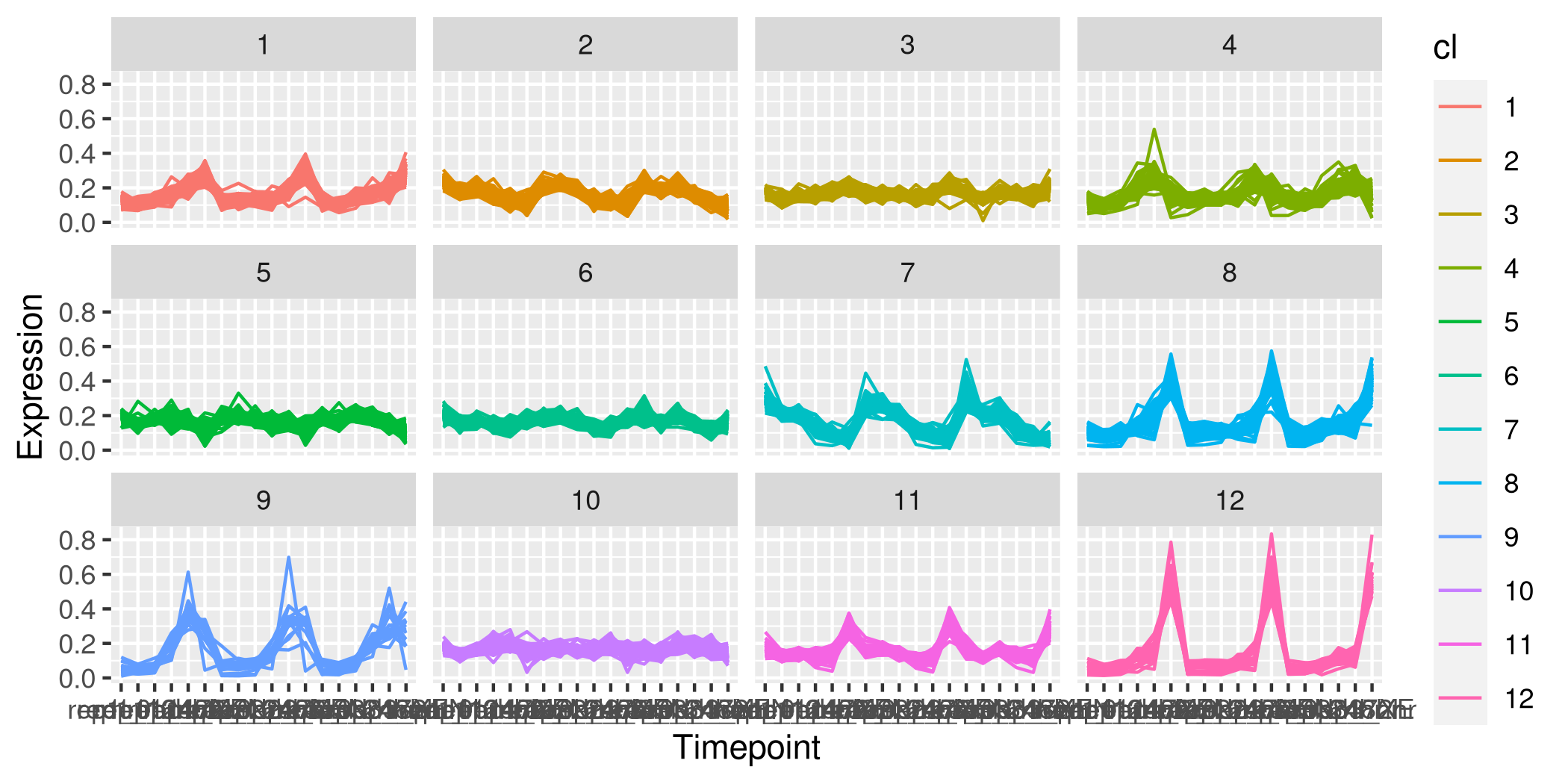
我希望可視化看起來像這樣,樣本在 x 軸上,運算式在 y 軸上。這些圖看起來像是facet_wrap()在 ggplot 中生成的:
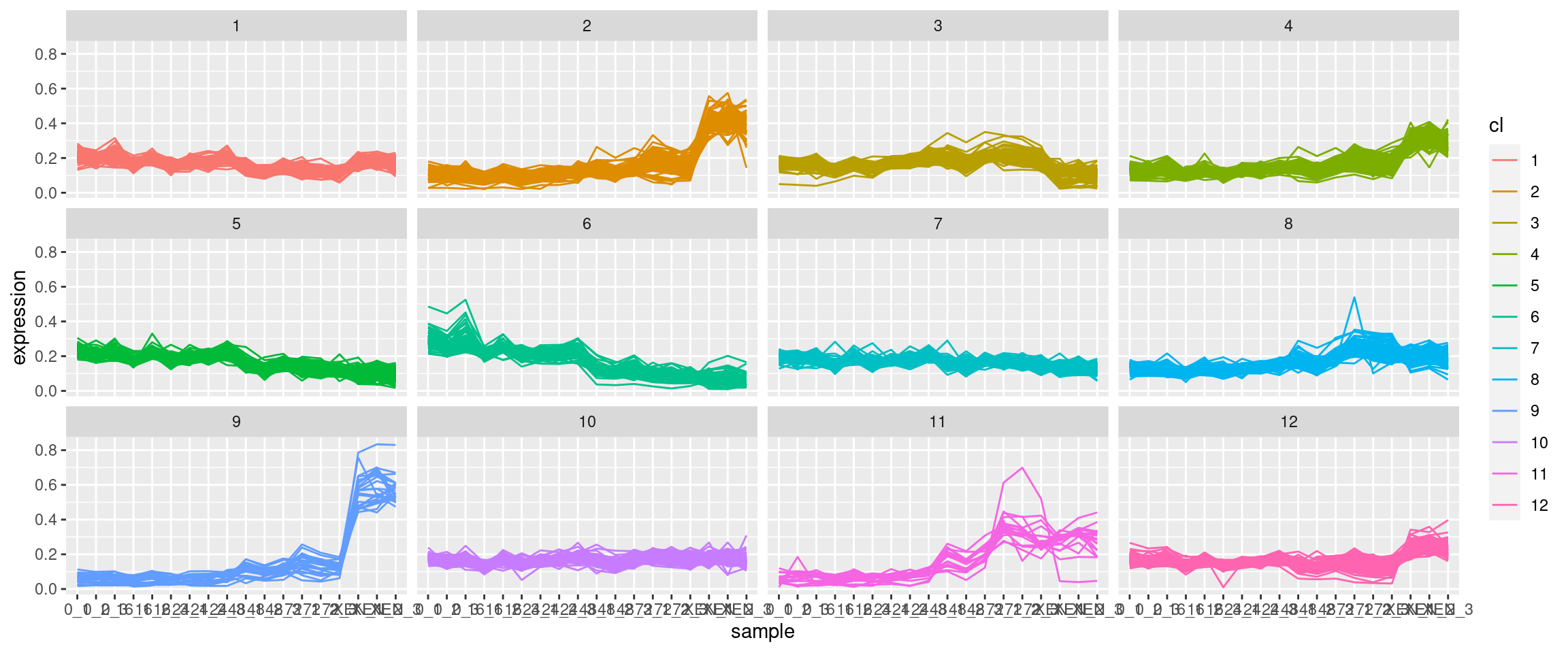
對于 ggplot,需要將資料作為資料框提供,其中有一列用于表示單個蛋白質的集群身份。此外,資料需要采用長格式。我嘗試旋轉(pivot_longer)原始資料集,但當然有大量的資料點。此外,我發布的影像顯示,對于任何一個圖,彩色線的數量都小于蛋白質的總數,這表明可能首先對資料集進行了降維,但我不確定。到目前為止,我一直在運行沒有降維的 kmeans 演算法。我可以就如何制作這個情節獲得指導嗎?
uj5u.com熱心網友回復:
這是我對情節進行逆向工程的嘗試:
library(pRolocdata)
library(dplyr)
library(tidyverse)
library(magrittr)
library(ggplot2)
mulvey2015 %>%
Biobase::assayData() %>%
magrittr::extract2("exprs") %>%
data.frame(check.names = FALSE) %>%
tibble::rownames_to_column("prot_id") %>%
mutate(.,
cl = kmeans(select(., -prot_id),
centers = 12,
nstart = 10) %>%
magrittr::extract2("cluster") %>%
as.factor()) %>%
pivot_longer(cols = !c(prot_id, cl),
names_to = "Timepoint",
values_to = "Expression") %>%
ggplot(aes(x = Timepoint, y = Expression, color = cl))
geom_line(aes(group = prot_id))
facet_wrap(~ cl, ncol = 4)
至于您的問題,pivot_longer除非它無法找到鍵中的唯一組合或與資料型別轉換相關的問題,否則通常會非常高效。情節可以通過以下方式改進:
- 調整
alpha引數geom_lines(例如 alpha = 0.5),以提供線條密度的概念 - 找到一個好的縮寫和順序
Timepoint - 改變axis.text.x方向
uj5u.com熱心網友回復:
這是我自己的,與上述非常相似的解決方案。
dfsa_mul <- data.frame(scale(assay(mul)))
dfsa_mul2 <- rownames_to_column(dfsa_mul, "protID")
將 kmeans$cluster列添加到dfsa_mul2資料框中。執行后只更改clus為一個因子pivot_longer
dfsa_mul2$clus <- ksa_mul$cluster
dfsa_mul2 %>%
pivot_longer(cols = -c("protID", "clus"),
names_to = "samples",
values_to = "expression") %>%
ggplot(aes(x = samples, y = expression, colour = factor(clus)))
geom_line(aes(group = protID))
facet_wrap(~ factor(clus))
這會生成一系列與@sbarbit 發布的圖表相同的圖表。
轉載請註明出處,本文鏈接:https://www.uj5u.com/qianduan/436081.html
上一篇:在多個圖的頂部添加標題