我真的希望你有時間在這里幫忙。所以,我試圖做一個我想洗掉默認圖例的情節,只是因為它沒有任何意義,因為它基于 0-1 的大小(我嘗試使用 legend.position = "無”,但后來我想添加一個根據我的情節制作的新圖例,這樣我就有三個選項:小點(log fold < 2)、藍點(p < 0.001 和 log fold < 2) ,和紅點(p < 0.001 和 log fold > 2)根據我的圖表。但是我無法洗掉默認圖例并仍然添加新圖例?!
提前非常感謝!
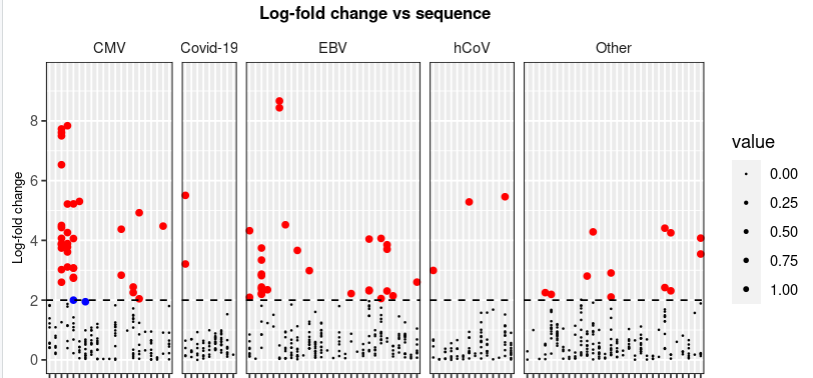
下面是我繪制圖表的代碼..
maximum_y <- my_data_clean_aug %>%
pull(log_fold_change) %>%
max() %>%
round() 0.5
#Determining the numbers of sequences per virus strain (Origin) and setting a threshold.
threshold <- my_data_clean_aug %>%
count(Origin) %>%
filter(n > 50) %>%
count() %>%
pull()
#Pooling all groups of vira with less than 50 hits into HHV or Others
my_data_clean_aug_pooling <- my_data_clean_aug %>%
mutate(Origin = as.factor(Origin)) %>%
mutate(newID = fct_lump(Origin, threshold)) %>%
mutate(value = case_when(log_fold_change <= 2 ~ 0,
0.001 < p & log_fold_change >= 2 ~ 0,
0.001 >= p & log_fold_change >= 2 ~ 1))
pointsofinterest <- my_data_clean_aug_pooling %>%
filter(0.001 >= p & log_fold_change >= 2)
pointswithpsig <- my_data_clean_aug_pooling %>%
filter(0.001 >= p & log_fold_change < 2)
my_data_clean_aug_pooling %>%
ggplot(aes(x = Peptide,
y = log_fold_change))
facet_grid(.~newID,
scales = "free_x",
space = "free")
geom_point(aes_string(size = "value"))
geom_point(data = pointsofinterest,
color = "red")
geom_point(data = pointswithpsig,
color = "blue")
geom_hline(yintercept = 2,
linetype = "dashed")
scale_y_continuous(limits = c(0,
maximum_y),
breaks = seq(0,
maximum_y,
2))
theme(plot.title = element_text(size = 10,
hjust = 0.5,
face = "bold"),
axis.text.x = element_text(size = 5,
angle = 90,
vjust = 0.5,
hjust = 1),
strip.background = element_rect(fill = "white"),
panel.border = element_rect(colour = "black",
fill = NA),
axis.title.x = element_text(size = 8),
axis.title.y = element_text(size = 8),
plot.background = element_rect(fill = "transparent",
color = NA))
labs(x = "ID",
y = "Log-fold change",
title = "Log-fold change vs sequence")
scale_size(range = c(0.1,
1))
dput(head(my_data_clean_aug_pooling, 30))
structure(list(sample = c("BC372", "BC372", "BC372",
"BC372", "BC372", "BC372", "BC372", "BC372", "BC372", "BC372",
"BC372", "BC372", "BC372", "BC372", "BC372", "BC372", "BC372",
"BC372", "BC372", "BC372", "BC372", "BC372", "BC372", "BC372",
"BC372", "BC372", "BC372", "BC372", "BC372", "BC372"), log_fold_change = c(0.878480955476892,
0.0254158993036971, 0.169690374849339, 1.29365346670481, 0.950207146498172,
0.121582483746693, 0.29591552217522, 0.0493694708020405, 0.253196235065184,
0.511413610788978, 0.92777679529061, 0.633288220541381, 0.852617925189971,
0.245947820840199, 0.284143920808481, 0.54421651055215, 0.998865269852439,
0.468714806763581, 0.704136952532169, 0.334881411284732, 1.09989649348867,
0.44520995356178, 0.559300342753859, 0.198650181166743, 0.947415942094208,
0.0365273151532468, 0.129416762542994, 3.85327690599736, 0.912242173799338,
0.980016944958404), p = c(0.455815003793973, 0.9710277325421,
0.929138758106761, 0.106508575848957, 0.325186030411862, 0.933801784951691,
0.929138758106761, 0.96549305958931, 0.929138758106761, 0.776892782297412,
0.325186030411862, 0.635666815285353, 0.382558882746048, 0.929138758106761,
0.929138758106761, 0.722931599232632, 0.325186030411862, 0.815874297477519,
0.529382980477629, 0.929138758106761, 0.238130758200615, 0.827129935665299,
0.711217028978768, 0.929138758106761, 0.325186030411862, 0.96549305958931,
0.933280410383701, 2.15547277536054e-13, 0.349668295725122, 0.325186030411862
), HLA = c("A0201", "A0201", "A0201", "A0201", "A0201", "A0201",
"A0201", "A0201", "A0201", "A0201", "A0201", "A0201", "A0201",
"A0201", "A0201", "A0201", "A0201", "A0301", "A0301", "A0301",
"A2402", "A2402", "A2402", "A2402", "B0702", "B0702", "B0702",
"B0801", "B0801", "B0801"), Origin = structure(c(5L, 1L, 5L,
9L, 19L, 5L, 7L, 18L, 1L, 3L, 5L, 14L, 14L, 9L, 5L, 3L, 5L, 5L,
5L, 3L, 3L, 2L, 14L, 5L, 3L, 15L, 5L, 5L, 3L, 3L), .Label = c("B19",
"BKPyV", "CMV", "Covid-19", "EBV", "FLU-A", "HAdV-C", "hCoV",
"HHV-1", "HHV-2", "HHV-6B", "HIV-1", "HMPV", "HPV", "JCPyV",
"NWV", "unknown", "VACV", "VZV"), class = "factor"), Peptide = c("v16",
"a47", "a49", "a50", "a51", "a52", "a53", "a55", "a57", "a58",
"a59", "a60", "a61", "a64", "a65", "a66", "a67", "v18", "v25",
"a68", "a74", "a77", "a80", "a81", "v14", "a87", "a89", "v17",
"v22", "a90"), newID = structure(c(3L, 5L, 3L, 5L,
5L, 3L, 5L, 5L, 5L, 1L, 3L, 5L, 5L, 5L, 3L, 1L, 3L, 3L, 3L, 1L,
1L, 5L, 5L, 3L, 1L, 5L, 3L, 3L, 1L, 1L), .Label = c("CMV", "Covid-19",
"EBV", "hCoV", "Other"), class = "factor"), value = c(0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 1, 0, 0)), row.names = c(NA, -30L), class = c("tbl_df",
"tbl", "data.frame"))
uj5u.com熱心網友回復:
這是一種方法。
與其將資料過濾成 3 個不同的資料集,不如創建一個新列,其值對應于多個條件。我已經呼叫了新變數Colour,但任何有意義的名稱都可以。這些值是通過一個陳述句分配的case_when,默認值為"black". 然后以比例手動給出顏色。
要洗掉value圖例,它現在使用引數guide。
我還定義了一個 custom theme,以使問題代碼更清晰。
theme_LasseVoss <- function(){
theme_minimal() % replace%
theme(
plot.title = element_text(size = 10,
hjust = 0.5,
face = "bold"),
axis.text.x = element_text(size = 5,
angle = 90,
vjust = 0.5,
hjust = 1),
strip.background = element_rect(fill = "white"),
panel.border = element_rect(colour = "black",
fill = NA),
axis.title.x = element_text(size = 8),
axis.title.y = element_text(size = 8),
plot.background = element_rect(fill = "transparent",
color = NA)
)
}
maximum_y <- ceiling(max(my_data_clean_aug_pooling$log_fold_change))
my_data_clean_aug_pooling %>%
mutate(Colour = case_when(
0.001 >= p & log_fold_change >= 2 ~ "interest",
0.001 >= p & log_fold_change < 2 ~ "psig",
TRUE ~ "other"
)) %>%
ggplot(aes(x = Peptide, y = log_fold_change))
geom_point(aes(size = value, colour = Colour))
geom_hline(yintercept = 2, linetype = "dashed")
#
scale_color_manual(
name = "Colour",
values = c(other = "black", interest = "red", psig = "blue")
)
scale_size(
range = c(0.1, 1),
guide = "none"
)
scale_y_continuous(
limits = c(0, maximum_y),
breaks = seq(0, maximum_y, 2)
)
#
labs(x = "ID", y = "Log-fold change",
title = "Log-fold change vs sequence")
facet_grid(.~newID, scales = "free_x", space = "free")
theme_LasseVoss()
uj5u.com熱心網友回復:
通常,如果您避免單獨繪制資料的子集,則更容易使圖例正常作業,因此獲得繪圖的一種方法是添加一個額外的列,指示它是哪種型別的點。請注意,示例資料不包括重要點。然后,您可以將代碼更改為:
my_data_clean_aug_pooling <- my_data_clean_aug_pooling %>%
mutate(group = factor(ifelse(0.001 >= p, ifelse(log_fold_change >= 2, "Interesting", "Significant"), "Boring"),
levels=c("Boring", "Interesting", "Significant")))
levels(my_data_clean_aug_pooling$group)
#[1] "Boring" "Interesting" "Significant"
my_data_clean_aug_pooling %>%
ggplot(aes(x = Peptide,
y = log_fold_change, colour=group, size=group)
)
facet_grid(.~newID,
scales = "free_x",
space = "free")
geom_point()
geom_hline(yintercept = 2,
linetype = "dashed")
scale_y_continuous(limits = c(0,
maximum_y),
breaks = seq(0,
maximum_y,
2))
theme(plot.title = element_text(size = 10,
hjust = 0.5,
face = "bold"),
axis.text.x = element_text(size = 5,
angle = 90,
vjust = 0.5,
hjust = 1),
strip.background = element_rect(fill = "white"),
panel.border = element_rect(colour = "black",
fill = NA),
axis.title.x = element_text(size = 8),
axis.title.y = element_text(size = 8),
plot.background = element_rect(fill = "transparent",
color = NA))
labs(x = "ID",
y = "Log-fold change",
title = "Log-fold change vs sequence")
scale_colour_manual(values=c("Boring"="black","Interesting"="red","Significant"="blue"))
scale_size_manual(values=c("Boring"=0.1, "Interesting"=5, "Significant"=5))
這使:
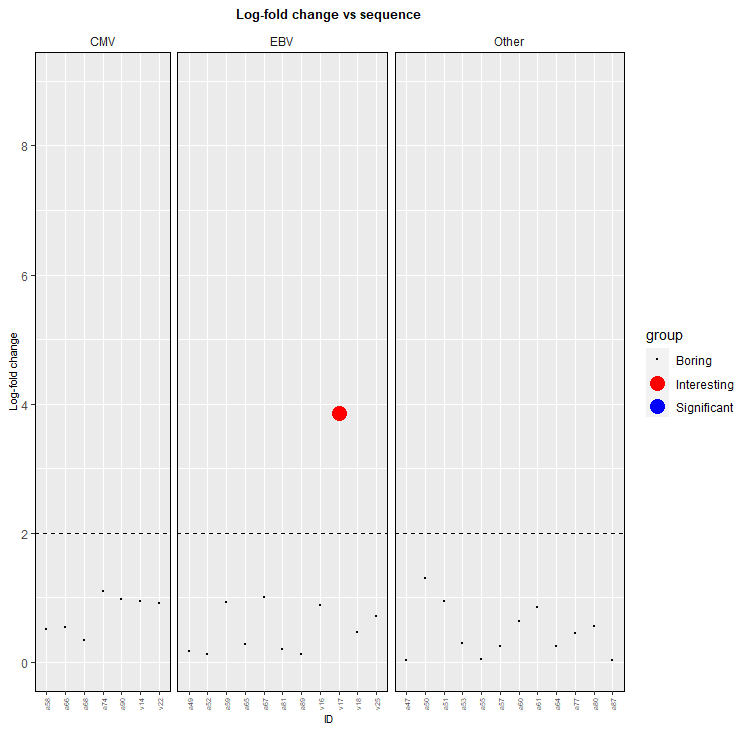
(我可能沒有為黑點選擇最好的名字!)
轉載請註明出處,本文鏈接:https://www.uj5u.com/yidong/471564.html