核苷酸序列(或 DNA 序列)通常由 4 個堿基組成: ATGC 為機器學習目的提供了一種非常好的、簡單和有效的編碼方式。
sequence = AAATGCC
ohe_sequence = [[1, 0, 0, 0], [1, 0, 0, 0], [1, 0, 0, 0], [0, 1, 0, 0], [0, 0, 1, 0], [0, 0, 0, 1], [0, 0, 0, 1]]
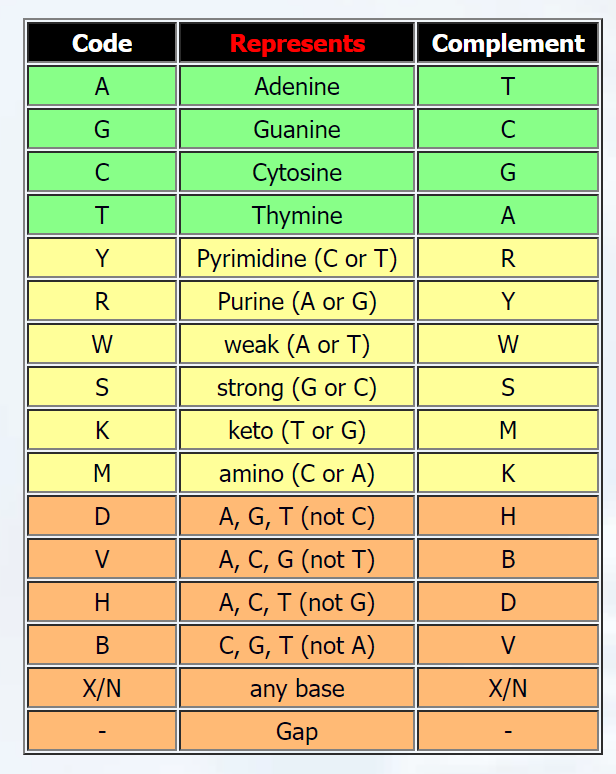
但是,當您考慮到 RNA 序列和有時可能在測序儀中出現的錯誤時,會添加字母 UYRWSKMDVHBXN……當單熱編碼時,您最終會得到一個 17 行的矩陣,其中最后 13 行通常都是0 的。
這是非常低效的,并且沒有賦予這些額外(模棱兩可)字母所具有的生物學意義。
例如:
- T 和 U 可以互換
- Y 代表有一個 C 或 T
- N 和 X 表示存在 4 個堿基中的任何一個(ATGC)
所以我制作了一本代表這種生物學意義的字典
nucleotide_dict = {'A': 'A', 'T':'T', 'U':'T', 'G':'G', 'C':'C', 'Y':['C', 'T'], 'R':['A', 'G'], 'W':['A', 'T'], 'S':['G', 'C'], 'K':['T', 'G'], 'M':['C', 'A'], 'D':['A', 'T', 'G'], 'V':['A', 'G', 'C'], 'H':['A', 'T', 'C'], 'B':['T', 'G', 'C'], 'X':['A', 'T', 'G', 'C'], 'N':['A', 'T', 'G', 'C']}
但我似乎無法弄清楚如何制作一個有效的單熱編碼腳本(或者是否有一種方法可以使用 scikit learn 模塊來做到這一點),它利用這個字典來獲得這樣的結果:
sequence = ANTUYCC
ohe_sequence = [[1, 0, 0, 0], [1, 1, 1, 1], [0, 1, 0, 0], [0, 1, 0, 0], [0, 1, 0, 1], [0, 0, 0, 1], [0, 0, 0, 1]]
# or even better:
ohe_sequence = [[1, 0, 0, 0], [0.25, 0.25, 0.25, 0.25], [0, 1, 0, 0], [0, 1, 0, 0], [0, 0.5, 0, 0.5], [0, 0, 0, 1], [0, 0, 0, 1]]
uj5u.com熱心網友回復:
這很有趣!我認為您可以使用具有適當值的字典來執行此操作。我添加了 scikit-learn 課程,因為您提到您正在使用它。見transform下文:
from sklearn.base import BaseEstimator, TransformerMixin
import numpy as np
nucleotide_dict = {
"A": [1, 0, 0, 0],
"G": [0, 1, 0, 0],
"C": [0, 0, 1, 0],
"T": [0, 0, 0, 1],
"U": [0, 0, 0, 1],
"Y": [0, 0, 1, 1],
"R": [1, 1, 0, 0],
"W": [1, 0, 0, 1],
"S": [0, 1, 1, 0],
"K": [0, 1, 0, 1],
"M": [1, 0, 1, 0],
"D": [1, 1, 0, 1],
"V": [1, 1, 1, 0],
"H": [1, 0, 1, 1],
"B": [0, 1, 1, 1],
"X": [1, 1, 1, 1],
"N": [1, 1, 1, 1],
"-": [0, 0, 0, 0],
}
class NucleotideEncoder(BaseEstimator, TransformerMixin):
def __init__(self, norm=True):
self.norm = norm
def fit(self, X, y=None):
return self
def transform(self, X, y=None):
f1 = lambda a: list(a)
f2 = lambda g: nucleotide_dict[g]
f3 = lambda c: list(map(f2, f1(c[0])))
f4 = lambda t: np.array(f3(t)) / np.sum(np.array(f3(t)), axis=1)[:, np.newaxis]
f = f3
if self.norm:
f = f4
return np.apply_along_axis(f, 1, X)
samples = np.array([["AAATGCC"], ["ANTUYCC"]])
print(NucleotideEncoder().fit_transform(samples))
輸出:
[[[1. 0. 0. 0. ]
[1. 0. 0. 0. ]
[1. 0. 0. 0. ]
[0. 0. 0. 1. ]
[0. 1. 0. 0. ]
[0. 0. 1. 0. ]
[0. 0. 1. 0. ]]
[[1. 0. 0. 0. ]
[0.25 0.25 0.25 0.25]
[0. 0. 0. 1. ]
[0. 0. 0. 1. ]
[0. 0. 0.5 0.5 ]
[0. 0. 1. 0. ]
[0. 0. 1. 0. ]]]
uj5u.com熱心網友回復:
我喜歡后一種方法,因為它更接近真正的含義:例如Y,不意味著C and T,而是C or T中的一個。如果沒有進一步的資訊,假設相等的概率(即權重)似乎是合理的。當然,這種非標準編碼需要通過損失函式的選擇來體現。
要回答您的問題:您可以預先計算從字母到編碼的映射,然后創建一個encode函式,該函式將 asequence作為輸入并將編碼序列作為-(len(sequence), 4)形回傳np.array,如下所示:
import numpy as np
nucleotide_dict = {'A':'A', 'T':'T', 'U':'T', 'G':'G', 'C':'C', 'Y':['C', 'T'], 'R':['A', 'G'], 'W':['A', 'T'], 'S':['G', 'C'], 'K':['T', 'G'], 'M':['C', 'A'], 'D':['A', 'T', 'G'], 'V':['A', 'G', 'C'], 'H':['A', 'T', 'C'], 'B':['T', 'G', 'C'], 'X':['A', 'T', 'G', 'C'], 'N':['A', 'T', 'G', 'C']}
index_mapper = {'A': 0, 'T': 1, 'G': 2, 'C': 3}
mapper_dict = dict()
for k, v in nucleotide_dict.items():
encoding = np.zeros(4)
p = 1 / len(v)
encoding[[index_mapper[i] for i in v]] = p
mapper_dict[k] = encoding
def encode(sequence):
return np.array([mapper_dict[s] for s in sequence])
這似乎產生了預期的結果,并且可能有些效率。
一個例子:
print(encode('AYSDX'))
列印以下內容:
array([[1. , 0. , 0. , 0. ],
[0. , 0.5 , 0. , 0.5 ],
[0. , 0. , 0.5 , 0.5 ],
[0.33333333, 0.33333333, 0.33333333, 0. ],
[0.25 , 0.25 , 0.25 , 0.25 ]])
uj5u.com熱心網友回復:
另一個允許不同長度序列的版本:
import numpy as np
nucleotide_dict = {
"A": [1, 0, 0, 0],
"G": [0, 1, 0, 0],
"C": [0, 0, 1, 0],
"T": [0, 0, 0, 1],
"U": [0, 0, 0, 1],
"Y": [0, 0, 1, 1],
"R": [1, 1, 0, 0],
"W": [1, 0, 0, 1],
"S": [0, 1, 1, 0],
"K": [0, 1, 0, 1],
"M": [1, 0, 1, 0],
"D": [1, 1, 0, 1],
"V": [1, 1, 1, 0],
"H": [1, 0, 1, 1],
"B": [0, 1, 1, 1],
"X": [1, 1, 1, 1],
"N": [1, 1, 1, 1],
"-": [0, 0, 0, 0],
}
norm = True
samples = np.array(["AAATGCC", "ANTUYCC", "".join(list(nucleotide_dict.keys()))[:-1]])
def nucleotide_encode(samples, norm=True):
m = map(list, samples)
f1 = lambda x: np.array(list(map(nucleotide_dict.get, x)))
f = f1
if norm:
f = lambda x: np.nan_to_num(f1(x) / np.sum(f1(x), axis=1)[:, np.newaxis])
return list(map(f, m))
for i, j in zip(samples, nucleotide_encode(samples, norm=norm)):
print(i)
print(j)
產量:
AAATGCC
[[1. 0. 0. 0.]
[1. 0. 0. 0.]
[1. 0. 0. 0.]
[0. 0. 0. 1.]
[0. 1. 0. 0.]
[0. 0. 1. 0.]
[0. 0. 1. 0.]]
ANTUYCC
[[1. 0. 0. 0. ]
[0.25 0.25 0.25 0.25]
[0. 0. 0. 1. ]
[0. 0. 0. 1. ]
[0. 0. 0.5 0.5 ]
[0. 0. 1. 0. ]
[0. 0. 1. 0. ]]
AGCTUYRWSKMDVHBXN
[[1. 0. 0. 0. ]
[0. 1. 0. 0. ]
[0. 0. 1. 0. ]
[0. 0. 0. 1. ]
[0. 0. 0. 1. ]
[0. 0. 0.5 0.5 ]
[0.5 0.5 0. 0. ]
[0.5 0. 0. 0.5 ]
[0. 0.5 0.5 0. ]
[0. 0.5 0. 0.5 ]
[0.5 0. 0.5 0. ]
[0.33333333 0.33333333 0. 0.33333333]
[0.33333333 0.33333333 0.33333333 0. ]
[0.33333333 0. 0.33333333 0.33333333]
[0. 0.33333333 0.33333333 0.33333333]
[0.25 0.25 0.25 0.25 ]
[0.25 0.25 0.25 0.25 ]]
轉載請註明出處,本文鏈接:https://www.uj5u.com/ruanti/462415.html
上一篇:選擇資料點鄰域來支持向量